

































~Authored By Anvi Gupta
Development and innovations in science happen at the interjection of different disciplines. And nothing would support this statement better than the Chemistry Nobel Prize-2024. This year’s Nobel Prize in chemistry has been awarded jointly to David Baker (University of Washington, Prize share: half), Damis Hassabis and John M. Jumper (Google Deep Mind, Joint Prize share: half). Baker has been awarded for computational protein design while Hassabis and Jumper have been recognized for protein structure prediction.
It is interesting to note that Damis Hassabis was a video game AI programmer and designer. An expert board games player who lent on to obtain his PhD in cognitive neuroscience from University College London. John Jumper, on other hand, obtained a Master of Philosophy in theoretical condensed matter physics. From the University of Cambridge and Doctor of Philosophy in theoretical chemistry from the University of Chicago in 2017. Together along with David Baker they have largely solved the problem of prediction of protein structure. Which has been a holy grail for biologists, biochemists and biophysicists for 50 grand years.

Why is protein structure prediction important?
Proteins are vital to almost all biological processes. Some proteins such as enzymes can speed up biochemical reactions within the body, some called structural proteins are responsible for cell integrity and overall shape. Some of them help in immune response while others can store nutrients or energy. We are familiar with a few of them such as the protein haemoglobin that transports oxygen in the body and that insulin that helps in absorption of glucose from blood. Thus, anything that affects and impacts protein production and its physiological functions will be very relevant to human health. Proteins are the biomolecules composed of building blocks called amino acids. There are 20 amino acids that combine to form various proteins.
The most important thing to understand is that the physiological function of any protein is dependent on its structure. Christian Anfinsen, an American scientist, who was awarded Nobel Prize in 1972 for his work on connection. Between the amino acid sequence and the biologically active conformation, got a protein structure to unfold and fold itself. He observed that the protein assumed exactly the same shape every time. This implies that the shape of a protein is determined by its sequence of amino acids. It opens the possibility of determining a protein’s 3-D structure if researchers know the sequence of amino acids that compose it. Still, the prediction of protein structure was an open problem for scientists for many decades but a significant one. Since as mentioned earlier, structure of the protein is the key to its functionality.
If researchers know the correlation between the structure adopted by a particular sequence of amino acids and the consequent properties (or functionalities), they can use that knowledge to design new synthetic proteins. In other words, it would provide a greater level of understanding of how a protein works. Which can allow us to create hypotheses about how to affect it, control it, or modify it. Unfortunately, solving the protein structure experimentally by conventional techniques like X-ray crystallography. Nuclear magnetic resonance and cryo- microscopy is highly efforts, time and cost intensive and may take months if not years. This is where the three Nobel laureates have contributed.
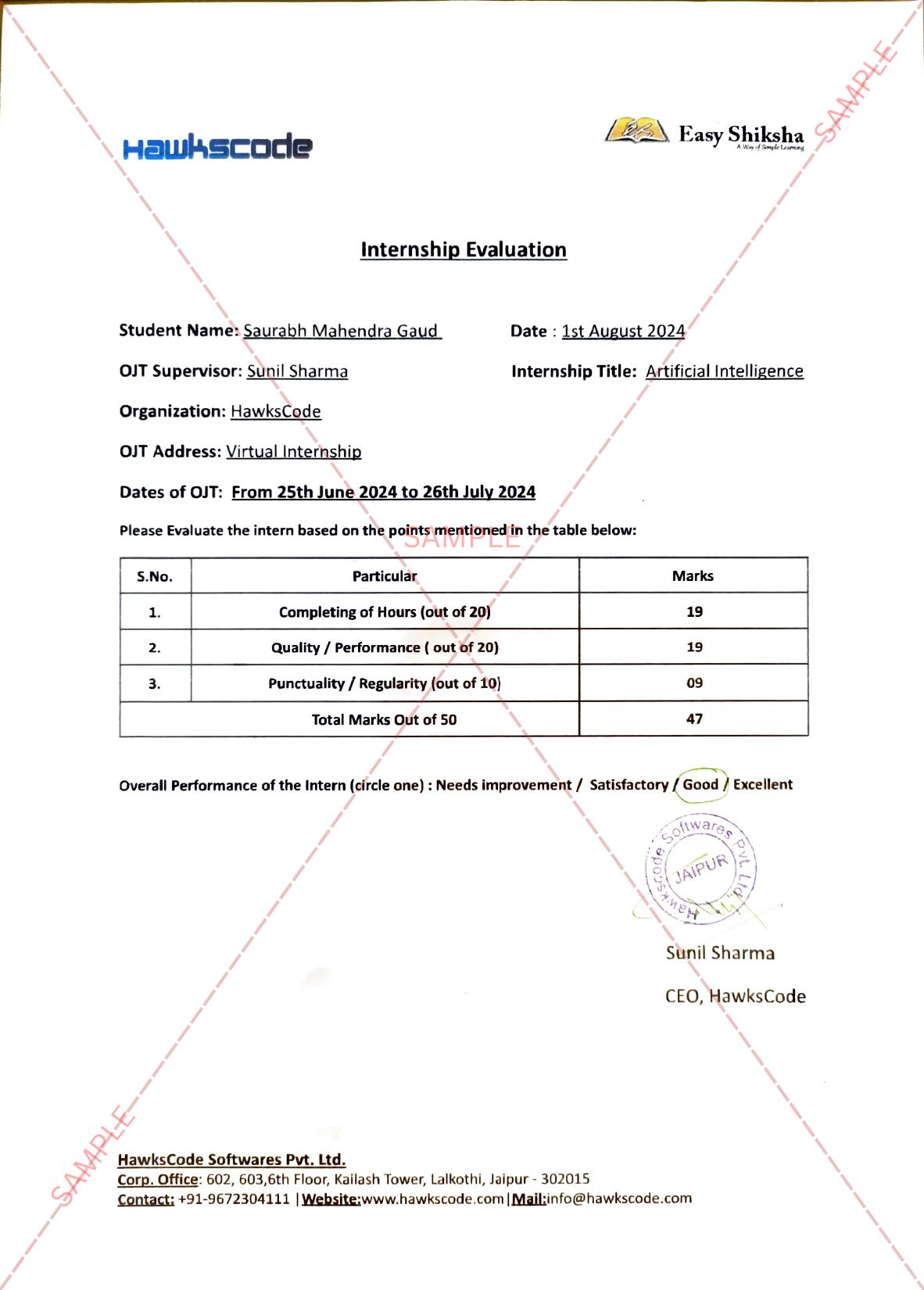
Important Announcement – EasyShiksha has now started Online Internship Program “Ab India Sikhega Ghar Se”
David Baker has developed a software called “Rosetta” in which if the protein structure is provided. One can get suggestions for possible amino acid sequences. This approach has helped researchers create entirely new synthetic proteins that do not exist in nature, with desired and improved functionalities that were previously unknown. This is significant because experimental methods take a long time.
Over the years, researchers have created a huge database of protein structures and the amino acid sequences that constitute them. Hassabis and Jumper developed the AI tool ‘AlphaFold’ and trained it on this data, enabling it to predict a protein’s structure with sufficient accuracy given its amino acids. They have upgraded it several times, and researchers worldwide now use it with a very high degree of success.
Together three of these, have made huge contributions to developing tools that make it easier and quicker to decode proteins’ structures, and in turn make entirely new proteins, in labs, feasible. This has far-reaching implications in medicine, for example, better understanding of diseases and drug design. In biotechnology for novel enzyme design), fast screening of drugs, protein engineering etc. As Nobel Committee puts it “That we can now so easily visualise the structure of these small molecular machines is mind boggling. It allows us to better understand how life functions, including why some diseases develop. How antibiotic resistance occurs or why some microbes can decompose plastic.”
Also Read: Allen Students bags 4 gold medals in IJSO
With platforms like Easyshiksha, the possibilities are endless. Start exploring the world of distance learning today and unlock your potential!





















{kind=link}
{kind=link}
{kind=link}